Dog’s DNA methylome uncovers hints on human cancer metastasis
Dog’s DNA methylome uncovers hints on human cancer metastasis

Tracking back human civilization there are evidences of dog domestication as far as 10,000 years ago. During this time, the ancient wolves’ genome evolved to give rise to the domestic dog that gradually adapted to human habitat as many organic functions were selected in detriment of others. Dogs have similar circadian rhythms, have adapted to human diet, and are often considered as family members. Hence, as a consequence of being exposed to the same environmental pressure,dogs and humans have developed similar physiological and adaptive responses.
Animal models have been used in biomedical research for many years given their resemblance to humans. They mimic common alterations occurring in human disease, and given the resemblance these have been key in understanding the pathobiology, as well as to study the etiology, prevention and therapy of human cancer and other disorders including diabetes or spinal cord injury, among others. Specifically, canine models have been extensively used since are considered to be closer to humans than rodents regarding physiology and metabolism 12. The sequencing of the dog´s genome in 2005 3 provided the molecular basis for the so-long evidenced parallelism with human disease.
In 1958 researchers S.H. Madin and N.B. Darby established an immortal cell line from a normal adult female cocker spaniel (MDCK cell line) that has afterwards been extensively used by the research community in a variety of contexts. Specifically, these cells were found to undergo epithelial-to-mesenchymal transition (EMT) upon exposure to the TGF-beta cytokine, a morphological change in cells that parallels the behavior of cancer cells as they leave the primary tumor and invade distant organs to give rise to metastases. The process of EMT takes place during embryonic development and involves migration of precursor cells to form the tissues and organs of growing organisms. Also, in adult organisms EMT naturally occurs during the process of wound-healing where epithelial cells experience phenotypic changes that allowfortissue repair. At the molecular level, this transition from an epithelial static state into a dynamic mesenchymal phase is tightly regulated through the coordinated action epigenetic players that dictate the activity of the genome in response to the cellular context. In human cancer, this molecular control is disrupted and aberrant changes in the conformation of the chromatin alter gene expression promoting the dissemination of cancer cells to other organs. For this reason, it is of great importance to understand the factors that allow cells to escape the primary tumor and invade distant organs.
Several models have been established to study EMT in vitro. Among them, the Madin-Darby canine kidney cell line (MDCK) has been broadly used to explore the molecular events occurring during EMT since it greatly resembles the metastatic dissemination of cancer cells in epithelial cancers 4; yet the molecular changes governing this process have only been partially devised. Recently, the laboratory of Dr Esteller in Barcelona has uncovered a series of epigenetic modifications on the DNA that are actively involved on this phenotypic transition in the canine cells, and provided some clues about the malignant EMT that causes metastasis in humans 5.
Epigenetics represents an additional regulatory layer that controls the activity of the genome without altering the nucleotide sequence. All the cells within an organism have the same DNA –genetics-, but the epigenetic –epi, from the Greek above- processes are the ones determining which genes are expressed at a particular time in a given cell type. Among these process, DNA methylationhas been the subject of most intense research on the epigenetics field given the wide amount of techniques available for its study. In a coordinated fashion along with histone modifications and other proteins that remodel the packaging of chromatin, the methylation of the DNA contributes to the epigenetic regulation of gene transcription in normal as well as in transformed tissues 6. Methyl groups are chemical tags that target the DNA at specific sequences, namely Cytosines (C) that are followed by Guanines (G) on the so called CG sites or dinucleotides. Moreover, these CG sites tend to cluster at the promoter sequences of around 60% of coding genes, configuring the CpG Islands – defined as the accumulation of CG sites at a frequency higher than it would be expected by mere probability. A plethora of reports over the last 20 years have well established that the methylation status of the CpG Island found in the vicinity – 2 kb around the transcription start site– of a gene promoter determines its transcriptional activity. High levels of DNA methylation stimulate the compacting of the chromatin around the promoter hence blocking the access of the transcription machinery; on the other hand, an unmethylated status reflects decondensed chromatin and active transcription (see Epigenetics takes us back to the Galápagos recently published in this blog) (Figure 1). Moreover, in contrast to genomic mutations epigenetic changes can be pharmacologically reversed by using demethylating agents that target aberrant DNA hypermethylation of the DNA.

Given the plasticity and reversibility of the process of EMT, it is more likely that the molecular changes triggering this transition are not caused by changes in the DNA sequence, but instead be a consequence of the reprogramming of the genome at an epigenetic level 7. On this study, Dr Esteller’s team aimed to describe the changes on DNA methylation that were associated with this epithelial-to-mesenchymal phenotype switch. To do so they compared the DNA methylation patterns of canine MDCK cells on their epithelial state and after inducing EMT through chronic treatment with TGF-beta. Thus, by performing whole-genome sequencing of epithelial and mesenchymal MDCK cells, authors compared both epigenomes at single-base resolution and spotted those DNA methylation changes that accompanied the process of EMT (Figure 2). This approach allowed researchers to describe the changes occurring on a simple cellular model that could be then extrapolated to uncover similar defects triggering breast cancer progression (5).

Researchers found genes that had been previously described to be epigenetically silenced –hypermethylated- during EMT, but also found new ones whose expression was altered as a consequence of changes in DNA methylation on their promoters. Moreover, when EMT was no longer induced (upon TGF-beta withdrawal from the culture media), cells progressively recovered their initial epithelial morphology, and so did the DNA methylation levels of the identified genes, reflecting the cell plasticity associated to EMT. Interestingly, this study also uncovered how the reshuffling of methyl groups along the gene bodies –exons and introns- had also a deep impact on gene expression, but in an opposed manner as promoter DNA methylation does (Figure 3). This observation is in agreement with the so called DNA methylation paradox 8, whereby genes actively transcribed require unmethylated promoters but high methylation levels at the gene bodies to trigger transcriptional elongation that culminates with the production of mRNAs. This and related works are suggesting that DNA methylation is a more dynamic process than previously anticipated, and far from being the endproduct of the action of upstream epigenetic regulators, it also has an active role in defining the activity of the genome and ultimately the functional state of the cell.
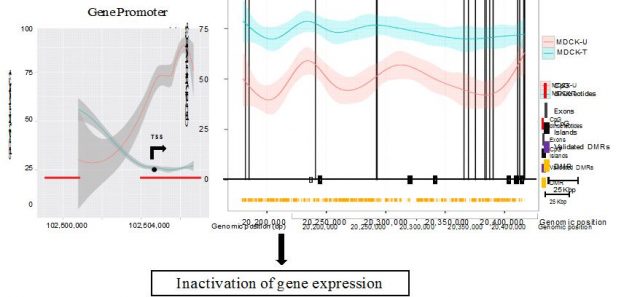
Besides, despite the fact that MDCK cells are non-transformed –were not derived from a tumor sample-, they recapitulate some of the alterations that occur during cancer EMT. Consequently, authors asked whether any of the changes identified on the canine model where recapitulated by human cancer cells. When researchers looked at the same genes on human breast cancer cells undergoing metastatic EMT, they found that many of them were equally altered. This indicates that there are epigenetic mechanisms that have been evolutionarily conserved and influence the morphology of cells when the physiological context –either during development or as part of a neoplastic process- requires so. Moreover, the great resemblance between the human and canine epigenomes supports previous evidences of parallel evolution where both human and dogs have taken similar paths to adapt to environmental cues.
At this point many exciting questions require to be addressed. Could EMT be reversed by using pharmacological inhibition of DNA methylation changes? How do the promoter and gene body DNA methylation status determine the transcription of a coding gene? What is the role of the epigenetic factors in the evolutionary response to a changing environment? The implementation of these high-throughput techniques allow researchers toread and interpret the information encoded on the genome, and is providing with exciting new clues about the regulation of the genome in diverse contexts. It therefore seems that the human’s best –animal- friend, besides lying by our bed and being faithful companions, can also give us hints on cellular plasticity, epigenetics and cancer.
References
- Ostrander EA. Both ends of the leash–the human links to good dogs with bad genes. N Engl J Med. 2012 Aug 16; 367(7):636-46. ↩
- Khanna C, Lindblad-Toh K, Vail D, London C, Bergman P, Barber L, et al. The dog as a cancer model. Nat Biotechnol. 2006 Sep; 24(9):1065-6. ↩
- Lindblad-Toh K, Wade CM, Mikkelsen TS, Karlsson EK, Jaffe DB, Kamal M, et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature. 2005 Dec 8;438(7069):803-19. ↩
- Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014 Mar; 15(3):178-96. ↩
- Carmona F.J., E. Vidal, A. Gomez, H. Heyn, Y. Hashimoto, M. Vizoso, A. Martinez-Cardus, S. Sayols, H. J. Ferreira & J. V. Sanchez-Mut & (2014). A Comprehensive DNA Methylation Profile of Epithelial-to-Mesenchymal Transition, Cancer Research, 74 (19) 5608-5619. DOI: http://dx.doi.org/10.1158/0008-5472.can-13-3659 ↩
- Bergman Y, Cedar H. DNA methylation dynamics in health and disease. Nat Struct Mol Biol. 2013 Mar; 20(3):274-81. ↩
- Wai Leong Tam& Robert A Weinberg. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nature Medicine 2013 19, 1438–1449 ↩
- Jones PA. The DNA methylation paradox. Trends Genet. 1999 Jan; 15(1):34-7. ↩