σ-hole bond as a preliminary stage of SN2 reaction
σ-hole bond as a preliminary stage of SN2 reaction
The SN2 reaction is one of the most important processes in organic chemistry and has been the subject of numerous experimental and theoretical investigations 12. The designation SN2 stands for substitution nucleophilic bimolecular. The reaction’s equation, X + R-Y → X-R + Y, summarizes bond formation by the attacking nucleophile X with the moiety R and concerted bond cleavage of the substituted leaving group Y. If the substrate under nucleophilic attack is chiral the SN2 mechanism proceeds with inversion of stereochemistry at the central R atom. This inversion of configuration proceeds through transition state and it is called the Walden inversion. The carbon atom is usually considered as the centre (R) since the alkyl centers were mainly analyzed in SN2 reactions. However there are theoretical and experimental investigations where the other atoms of the 14th group of elements or even atoms of other groups like for example, nitrogen, phosphorus or boron play the role of centers in SN2 reactions.
In a case of alkyl centers the transition state of SN2 reaction is characterized by a pentacoordinated carbon atom with a trigonal bipyramidal geometry, the departing and incoming groups occupying the apical positions. When the transition state is reached, the central carbon atom has gone from its initial sp3 hybridization to an sp2 state with an approximately perpendicular p orbital. One lobe of this p orbital overlaps with the nucleophile and the other with the leaving group. This is why a frontside SN2 mechanism has never been observed. The backside mechanism involves the maximum overlapping throughout the course of the reaction. During the transition state three nonreacting substituents and the central carbon are approximately coplanar. They are exactly coplanar if both the entering and leaving groups are the same.
There are different factors which govern the SN2 mechanism. One can mention steric effects, in practice, the mechanism does not occur at tertiary alkyl centers, solvent effects etc. It was found that the central atom properties may influence the significance of the steric effects. For example, the reversible addition reactions of methyl(halogeno)tin andmethyl(halogeno)germanium compounds to electron-rich platinium(II) complexes were analyzed; the kinetic and thermodynamic parameters have been obtained by variable temperature 1H NMR and UV-visible spectroscopic studies 3. It was found that methylhalotin species, Me3SnX (X = Cl, Br, I), react by an SN2 mechanism with a five-coordinated tin center in the transition state. For the germanium analogs, Me3GeCl, the variable temperature NMR spectral series shows the rate decrease of the SN2 reaction; the authors conclude that this is partly due to a size effect since for the smaller Ge-center than the Sn one the nucleophilic attack is less probable. This is why, for the related silicon complexes 1H NMR spectra have not shown any evidence of the SN2 reaction [3].
The analysis of numerous SN2 reactions and factors governing these processes leads to the question – how the reactants find the optimal arrangement to start orbital-orbital overlapping corresponding to the SN2 process? In other words – what are the preliminary stages of the SN2 reactions?
It was analyzed recently that the sp3 carbon centers can interact with electron rich centers (Y) via carbon 45. These interactions, C…Y, were named as the carbon bonds [5] and it was pointed out that they are characterized by the similar properties as the other intermolecular interactions such as for example the hydrogen bonds. The carbon bonds can play important role in numerous biological processes such as protein folding. The authors have stated that the SN2 reactions often involve a pentacoordinate C with two nucleophilic groups and that the C…Y interaction could play a stabilizing role in the intermediate to SN2 reactions [5]. Figure 1 presents the complex linked through the C…N carbon bond with the central pentacoordinated carbon atom.
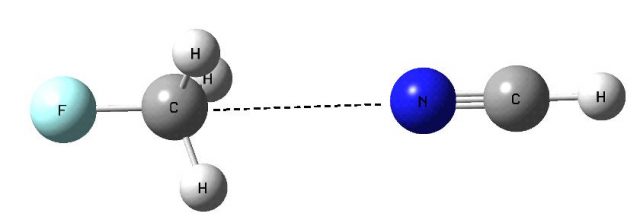
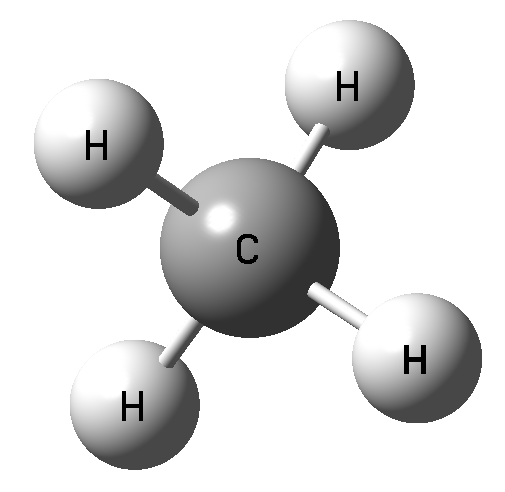
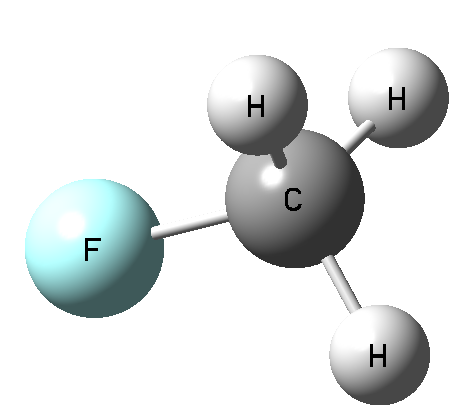
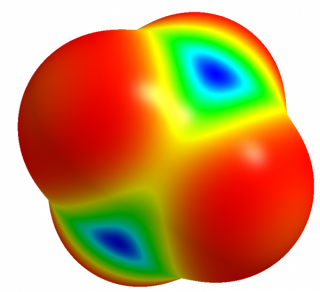
The interactions through the carbon atom and the other elements of the 14th group were analyzed [4] and it was stated that the carbon may act as the Lewis acid center interacting with Lewis bases if it is characterized by the sites of the positive electrostatic potential (EP). This is why the C…Y interactions are not observed in a case of methane (also SN2 reactions for methane hardly proceed) since the carbon center possess the negative electrostatic potential (Fig. 2) and can not interact with nucleophiles. On the other hand there is the positive EP site at C-atom for the CFH3 molecule in the elongation of F-C bond. This site is the result of the polarization of F-C bond and consequently the depletion of the electron density in the F-C σ-orbital. This is why such depletions along the bonds are named σ-holes and the corresponding intermolecular interactions are named as the σ-hole bonds; indeed the C…Y interactions analyzed here are the links through the σ-holes.
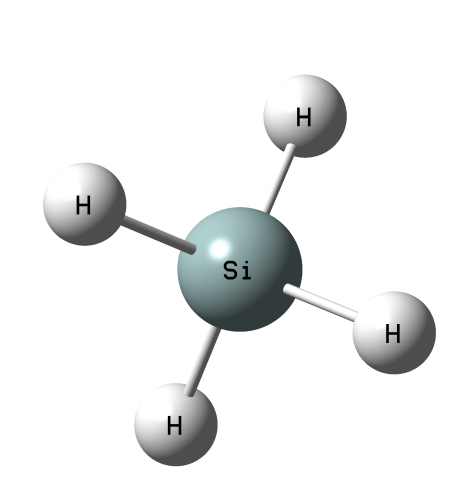
The positive EP sites for the atoms of the 14th group (tetrels) and the corresponding σ-hole bonds (tetrel bonds) were analyzed recently 6. It was found that the electrostatic potential at the tetrel atom increases if its atomic number increases. For example, there is the positive electrostatic potential at the Si-atom for the SiH4 molecule (Fig. 3) thus the interactions of this Si-center with Lewis bases are possible. As it was mentioned above, the latter interaction is not observed for methane. The σ-hole concept partly explains the experimental observations on reactions of methylhalotin species and their germanium and silicon analogs presented earlier here, the SN2 mechanism detected for Sn species and next for Ge analogs (the rate decrease of the SN2 reaction for Ge species) are not observed for Si moieties since the positive electrostatic potential at the central reaction site decreases in the following order Sn > Ge > Si > C. Theoretically the Si-atom may act as the Lewis acid center, however for the methylhalosilicon moieties there are large substituents thus their hindering influence takes place. However one can state that the tetrel bond or particularly carbon bond may be treated as the initiation of the SN2 process.
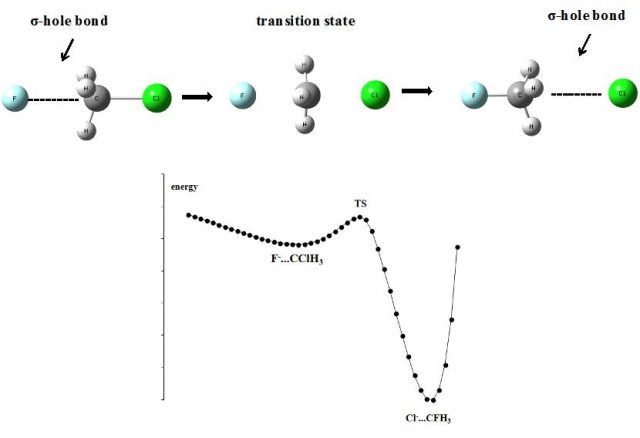
Figure 4 presents the scheme of the SN2 reaction, F– + CH3Cl → Cl– + CH3F, where the reactants and products form σ-hole bonds. This process may be described in the following way in terms of the σ-hole concept. The CClH3 molecule is characterized by the positive electrostatic potential at the carbon atom in the elongation of the Cl-C bond. This positive EP site exists owing to the polarization of Cl-C bond and consequently the depletion of the electron density at the σ-bond at the C-atom. This is why the carbon atom as the Lewis acid center interacts with the F– Lewis base forming the F–…C carbon bond (σ-hole bond). This complex corresponds to the shallow potential energy minimum close to the transition state (TS) of the SN2 reaction (Fig. 4). There is the electron charge shift from F– to the CClH3 species and next from C-atom to the Cl-substituent leading to the strong polarization of C-Cl bond and strong attraction between F– and C what further results in the electron charge redistribution corresponding to the TS structure. For TS the molecular structure close to the trigonal bipyramid is observed with the pentacoordinate carbon atom and with the planar CH3 group. The F–…C and Cl–…C contacts are perpendicular to the CH3 plane and the formal charge of chlorine and fluorine is situated between 0 and -1. Next, the strong polarization of Cl-C bond results in the formation of the Cl– + CH3F product corresponding to the global potential energy minimum where also the σ-hole bond is observed, i.e. Cl–…C carbon bond.
Numerous σ-hole bonds were analyzed recently, i.e. carbon bonds [5], tetrel bonds [4,6], as well as the N…N, N…P and N…As interactions 7 and it was found that many of them may be treated as the preliminary stage of the SN2 reaction. This is the most important that for them the reaction sites correspond to the extrema of the electrostatic potential surfaces, thus the initial steps of reactions are electrostatic in nature.
References
- J. March, Advanced Organic Chemistry, 4th ed., Wiley, New York, 1992. ↩
- A. Rauk, Orbital Interaction Theory of Organic Chemistry, 2nd ed., John Wiley & Sons, New York, 2001. ↩
- C.J.Levy, R.J.Puddephatt, J.Am.Chem.Soc. 1997, 119, 10127-10136. ↩
- A. Bundhun, P. Ramasami, J. S. Murray, P. Politzer J.Mol.Model. 2012, 19, 2739–2746. ↩
- D. Mani, E. Arunan, Phys.Chem.Chem.Phys. 2013, 15, 14377–14383. ↩
- Grabowski S.J. (2014). Tetrel bond–σ-hole bond as a preliminary stage of the SN2 reaction, Physical Chemistry Chemical Physics, 16 (5) 1824. DOI: 10.1039/c3cp53369g ↩
- S.J. Grabowski, Chem.–Eur. J. 2013, 19, 14600–14611. ↩
1 comment
[…] aurkitzen dute erreaktiboek erreakziorako neurririk egokiena? Slawomir Grabowskik azaltzen digu σ-hole bond as a preliminary stage of SN2 reaction […]