DFT insights into bond-breaking processes in photoresponsive ruthenium drugs
DFT insights into bond-breaking processes in photoresponsive ruthenium drugs
Light can do more than illuminate matter. In some metal complexes, it can break chemical bonds in a highly controlled way, releasing specific molecules only when and where light is applied. This idea lies behind photoactivated chemotherapy, a strategy in which relatively inactive compounds become chemically reactive after irradiation. Ruthenium complexes are among the most studied systems for this purpose because their excited electronic states can be carefully tuned through molecular design.
New research examined 1 a family of ruthenium compounds containing a terpyridine ligand and an acetonitrile molecule bound to the metal center. When these compounds absorb visible light, the ruthenium–acetonitrile bond can break, allowing the acetonitrile ligand to leave and another molecule, such as water, to take its place. Understanding exactly how this bond-breaking process occurs is important for designing more efficient photoresponsive drugs.
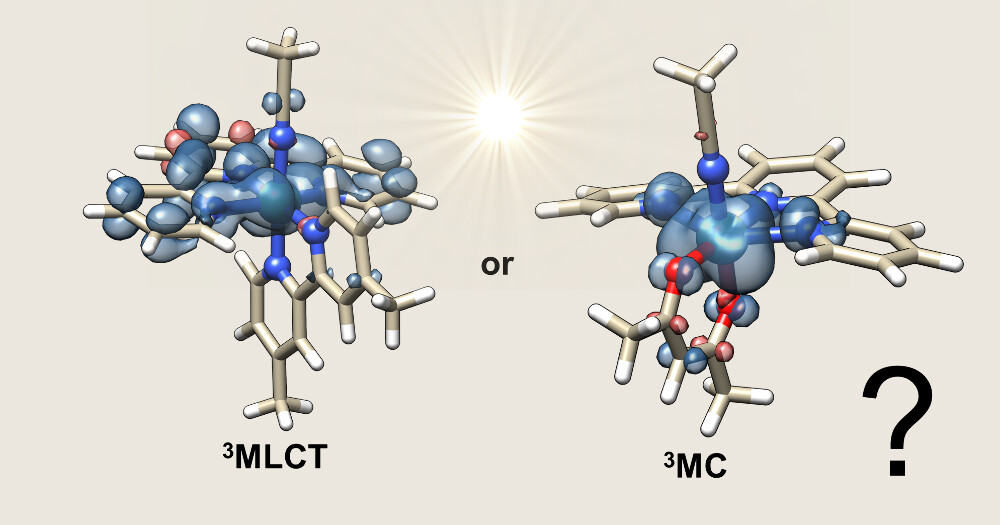
For many years, chemists have generally described the process using a sequence of excited states. After light absorption, the molecule undergoes an almost instantaneous process called intersystem crossing, landing in a triplet metal-to-ligand charge-transfer state, or 3MLCT for short. The superscript “3” denotes a triplet state, meaning the molecule carries two unpaired electrons after light absorption. In this state, one electron has been promoted from an orbital centered on the ruthenium atom into an orbital centered on the surrounding ligands. The system can then thermally convert (by overcoming an activation energy barrier) into another excited state called a metal-centered state, or 3MC. Traditionally, this 3MC state has been considered the key reactive state because it places electron density into orbitals that work against the metal–ligand bond, weakening it enough to trigger ligand loss.
However, experiments performed in recent years suggested that some ruthenium terpyridine complexes might not follow this classic picture. Surprisingly, compounds where the 3MC state was harder to reach energetically sometimes showed higher photodissociation efficiency. This raised the possibility that ligand release could occur directly from the 3MLCT state, bypassing the usual intermediate.
The study explored this question using density functional theory calculations. Several related ruthenium complexes were analyzed in water, including systems containing bipyridine-like ligands and others containing acetylacetonate ligands. The calculations reproduced experimentally measured trends very well, especially the energy barriers separating the 3MLCT and 3MC states.
One of the most important findings was that the reactive 3MC state looked very different from what chemists had expected. In many previous models, the metal–ligand bond destined to break becomes strongly elongated before dissociation occurs. Here, the ruthenium–acetonitrile bond lengthened only slightly. Instead, the major structural change involved distortion of the terpyridine ligand itself. The normally planar ligand bent out of shape, strongly perturbing the metal coordination environment.
This distortion turned out to be central to the reaction mechanism. As the terpyridine framework relaxed back toward planarity, acetonitrile dissociation became easier. The calculations identified transition states connecting the distorted 3MC structures to five-coordinate ruthenium intermediates in which acetonitrile had detached. The work therefore showed that ligand release can occur even without the strongly elongated metal–ligand geometries usually associated with dissociative excited states.
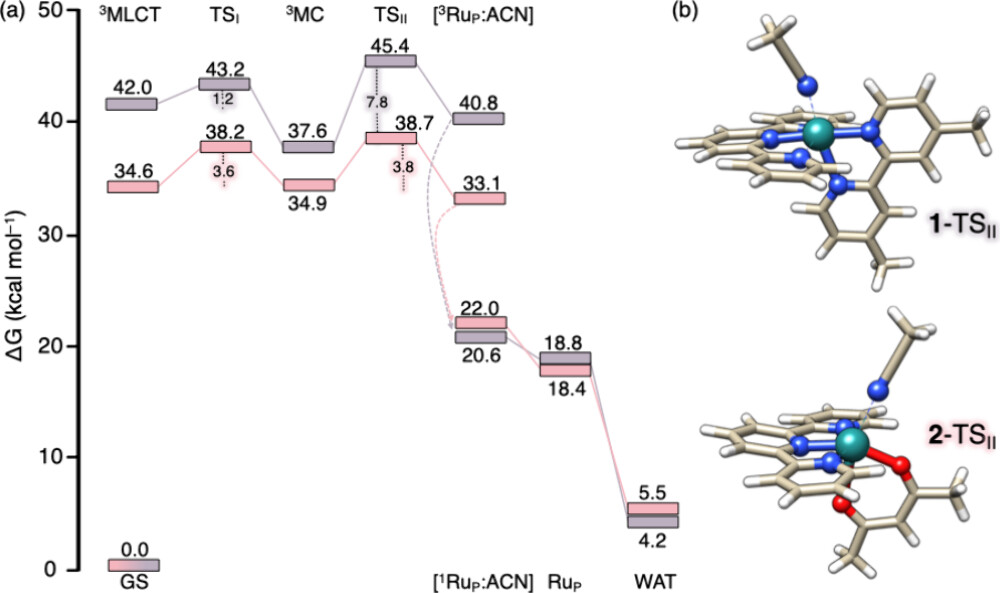
The results also clarified why different complexes display different photochemical efficiencies. In one complex, the conversion from 3MLCT to 3MC was relatively easy, but the subsequent acetonitrile release step required a larger energy barrier. In another complex, both processes had similar barriers. This means that the overall reaction rate is not determined solely by formation of the 3MC state. The actual bond-breaking step itself can become the limiting factor.
The calculations also tested the alternative idea of direct ligand loss from the 3MLCT state. Energy scans along the ruthenium–acetonitrile bond showed that this pathway is energetically plausible. For one complex in particular, the researchers succeeded in locating a transition state corresponding directly to acetonitrile dissociation from the 3MLCT state. This is significant because such a transition state had not previously been characterized computationally for ruthenium polypyridyl systems.
The study therefore suggests that the two mechanisms are not mutually exclusive. Depending on the structure of the complex, ligand release may occur through either a distorted 3MC state or directly from the 3MLCT state. In some compounds the two pathways may even compete simultaneously.
The work also explored how subtle structural modifications influence the photochemistry. Adding methyl groups at the ortho positions (closest to the metal-binding nitrogen atoms) on the terpyridine ligand introduced steric crowding that favored distortion of the ligand framework. This stabilized the distorted 3MC state and lowered the barrier for conversion from 3MLCT to 3MC, without greatly changing the energy required for acetonitrile release itself. Placing the methyl groups at neighbouring positions on the same ring had no such effect, confirming that the outcome depends precisely on where the modification is made. Such results provide a rational design strategy for controlling excited-state reactivity through molecular geometry.
Author: César Tomé López is a science writer and the editor of Mapping Ignorance
Disclaimer: Parts of this article may have been copied verbatim or almost verbatim from the referenced research paper/s.
References
- S. Scoditti, G. Mazzone, E. Sicilia, and L. Salassa (2026) DFT insights on ligand photodissociation pathways in ruthenium-terpyridine complexes: 3MLCT- or 3MC-triggered? Inorg. Chem. doi: 10.1021/acs.inorgchem.6c00971 ↩